bioconductor 中的注释系统
bioconductor 不仅提供了一系列的软件工具,而且还提供了包装好的注释信息来方便用户使用. 这些注释信息被分门别类的封装在数量众多的包package中.
用户可以在 bioconductor的BiocViews页面 中寻找需要的注释. 下图显示了在查询页面搜索 human的结果.
不用因为注释系统包括的包数量众多(图中显示962个),而担心学不过来.
这些包之间共用一系列公共的函数, 所以可以用同一套方法来从不同的包中查询数据.
而各个包之间差别更多的在于存放的数据内容以及作为搜索主键的类型.
共同的查询操作
我们用C.elegans 的注释包做例子,来了解一下使用bioconductor注释系统查询信息的通用操作.
首先我们载入这个包
library(org.Ce.eg.db)
载入之后, 我们可以得到一个和包同名的对象(object)用来查询数据. 这个对象可以被简单地类比为大型二维表格,
我们可以使用直接查询这个包同名对象的类型:
class(org.Ce.eg.db)
得到的结果是
[1] "OrgDb"
attr(,"package")
[1] "AnnotationDbi"
以上结果说明了 org.Ce.eg.db 这个包载入之后,内存中出现了一个名叫org.Ce.eg.db的对象. 这个 OrgDb类型的对象继承自 AnnotationDbi,
注释系统中的包的类型除了OrgDb之外,还有 ChipDb 和 TxDb类型.
虽然类型不同, 但都继承自AnnotationDbi . 所以 AnnotationDbi中columns, keytypes, keys select 这些函数在所有的包中都能用.
不同的包可能提供自己的函数, 但上述的四个函数完全通用, 并且查询信息足矣.
不同类型的包之间关联如下图所示:
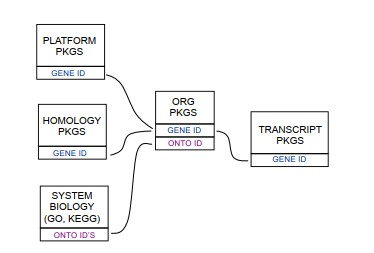
图中代表某类包的方框下彩色字 GENE ID 和 ONTO ID表示了主键类型.
查询通用步骤
我们继续使用二维表格的比喻来解释查询信息的过程. 比如一个m*n的二维表格 x, 有m行n列.
查询信息就是从表格中取部分行和部分列组成的子集.
可以分为三步:
- 确定主键所在的列,
- 用这个列中的部分元素来确定提取的部分行
- 确定需要提取的部分列.
对应到
keytypes(x)是用来说明x中哪些列可以用来查询.
keys(x,keytype, ...)就是返回keytype指定的某个列中所有的元素.
columns(x)就是用来说明x有哪些列的信息可以提取出来.
select(x, keys, columns, keytype, ...)在确定了查询的列keytype, 查询的值keys, 和输出的列columns之后, 提取需要的信息
由于数据可能存在冗余,不是所有的列都能用来查询,所以keytypes 和 columns 得到的结果不一定相同. 还有一个通用的函数 mapids, 这个函数类似select, 但是只提取单独的列.
bioconductor的注释包可以分成以基因为中心(gene centric)或者以基因组为中心(genome centric)
基因为中心的意思是,这个二维数据框中每一行代表一个基因. 基因组为中心表示每一行不是基因,而是基因组上的一个区域(特征).
org.Ce.eg.db 这个包是基因为中心的, 所以keys函数得到的结果是基因相关的ID.
比如
> head(keys(org.Ce.eg.db))
[1] "171590" "171591" "171592" "171593" "171594" "171595"
> head(keys(org.Ce.eg.db, keytype = "REFSEQ"))
[1] "NM_058260" "NP_490660" "NM_001306277" "NM_058259"
[5] "NP_001293206" "NP_490661"
默认参数的keys返回了entrez id, 而指定keytype为refseq id之后返回了同一批基因的refseq
id .
使用select 来查询信息的示例如下:
> select(org.Ce.eg.db, keys = head(keys(org.Ce.eg.db, keytype = "REFSEQ")), keytype = "REFSEQ", columns = c("REFSEQ", "ENTREZID", "GENENAME"))
REFSEQ ENTREZID GENENAME
1 NM_058260 171590 Alpha N-terminal protein methyltransferase 1
2 NP_490660 171590 Alpha N-terminal protein methyltransferase 1
3 NM_001306277 171591 Peptide P4
4 NM_058259 171591 Peptide P4
5 NP_001293206 171591 Peptide P4
6 NP_490661 171591 Peptide P4
这里, 我们使用keytype 为 REFSEQ, 主键的值依然不变, 输出的信息(columns)包含三列 REFSEQ, ENTREZID, GENENAME, 查询结果是一个包含上述三列信息的data.frame .
从注释包的名称看它所属类型
上文提到bioconductor的注释包可以分成基因中心和基因组中心, 考虑到生物信息学的应用场景, 这些注释包可以划分成更多的类别.
但我们可以从包的名称来直观的得知它们的类型.
基因组中心的包
基因组为中心的包中, 常见是转录本层面(transcriptome level)的注释.
这些包名称大致是 TxDb.xxxx.yyy.db 或者 EnsDb.xxxx.yy.db.
其中 xxxx 为物种名称, yyy 是版本.
包中数据的主键(每一行)是基因组上不同特征(feature)的区域(range), 这个区域对应某个特征,可能是转录本,可能是基因.
基因中心的包
而基因为中心的数据包涵盖的分类更多一些, 具体如下:
- 物种层面(Organism level)
这些数据库名称都是 org.<Ab>.<id>.db (比如 org.Sc.sgd.db),
<Ab> 是物种名称缩写, 比如Sc 代表了Saccharomyces cerevisiae, 注意大小写.
<id> 表明了主键的来源, 比如sgd 表示Saccharomyces Genome Database,而 eg 表示 Entrez Gene ids).
这些包载入后提供 OrgDb类型的对象.
- 平台层面(Platform level)
这些包往往用来注释芯片探针, 所以名称是 xxxx.db, 其中xxxx就是芯片的类型.
比如 hgu95av2.db,
这些包载入后提供的是ChipDb类型
- 同源性层面(Homology level)
这些包名称规律是 Hom.<Ab>.<xxx>.db
Hom 代表 homology, <Ab> 表示物种 , <xxx> 表示数据来源
- 系统生物学层面 (System-biology level)
这类注释包名字就很简单, 比如 GO.db 或者 KEGG.db