bioconductor 中的基础设施
很多人(包括我在内)开始接触bioconductor, 往往是先领了一个任务,比如差异表达分析要么是GEO数据挖掘之类. 网上搜索一遍得知可以使用R中的bioconductor来做, 如获至宝, 马不停蹄的照着网上的教程一边依样画葫芦来做分析,一边赌这些代码能够正常工作.
这样做的弊病就是如果教程上的代码运行如果出错,那么就一点都没法改. 表面上数据在代码中被一步步的处理直到最后结果, 但实际上我们并不知道这些代码背后是怎么运作的. 某一步出错之后,我们甚至不知道这个时候内存中的数据是什么样子,存放在哪. 所以了解一下bioconductor的基础数据结构和基本操作是很必要的, 至少代码出错的时候,能知道错在哪儿,方便搜索另一篇教程…..
然而bioconductor的数据结构稍微有些门槛,它是建立在S4类基础上,不同于base R的S3类.
而且它有自己的一套叙事方式, 各种乱七八糟的Range, View 之类的.
同时这些不同的数据结构之间往往共用很多泛型函数. 比如求宽度的width函数到处出现.
而bioconductor的文档属于手册性质, 更像是”懂”的人写给”懂”的人看得.
如果碰到问题的时候, 新人单看某个包的说明文档, 往往就容易被迷惑.
而bioconductor提供的功能相当灵活,多种方式可以得到同样结果,这让初学者更容易迷惑.
但是不管什么计算机语言,不管什么类库, 他们都是对客观实体的描述, 就好像一段代码的功能一定能用人话说明白. 所以这里,我们换个方向, 先讨论生物学基础引发的相关问题,然后再看bioconductor是如何解决这些问题的.
实际使用R的时候,总可以通过临时查阅帮助文档和试错来得到正确结果.关键是要能回忆起有这么一个概念或者功能. 因此接下来的讨论中虽然有一些代码,更值得关注的是代码对应的生物学 概念 和 操作. 我们假设读者知道基本的分子生物学,也对R有一部分了解, 比如能够用R来做回归分析.
一个生物问题
目前生物信息的研究基本围绕基因组(genome)和染色体(chromsome)展开. 基因是染色体上编码蛋白的功能区域,一个基因可能会包括多个转录本, 而一个转录本中包括多个外显子区域. 研究的生物学问题往往最终表现为这些区域(region)上的某些数值, 比如说某次测序的覆盖度. 除此以外我们也很关心这些区域附近的序列, 比如说两个外显子之间的内含子序列中的剪切位点信号.
假设我们现在要用计算机解答这么一个生物学问题: 外显子5’端的序列的保守性是否有别于基因其他区域?
问题中概念的转述
在开始回答这个问题之前, 不妨先用词性分析来看看这个问题中涉及了哪些概念和操作:
-
外显子(名词): 可以认为是染色体序列中最终对应肽链的区域, 那么5’端,也就是这个区域的一部分. 为了说明”区域”,我们要提前说明这个区域所出在的地方:染色体与其序列.
-
保守性(名词): 可以认为是与染色体序列一一对应的数据.为了解决这个问题,假设手头已经有了序列保守性的数据.
-
有别于(动词) 可以认为是保守性数值在不同区域之间的比较.
接下来把问题中涉及到的概念对应到bioconductor上来:
染色体上的区域
为了表示染色体上的区域,不妨把染色体抽象成一个一维向量, 这样,单个染色体上的区域只需记录起止点就足够表示 (对应IRanges)
而要表示有生物学意义的区域, 比如外显子exon, 那么在起止点之外,还要有染色体的信息, 以及 这个区域的其他一些生物学信息 (对应 GRanges)
一系列的exon可以用来表示一个转录本, 或者说区域组成的向量可以表示转录本. 那么为了表示基因这种包含多个转录本的概念, 我们需要使用list 来将这些区域的向量包括进去 (对应GrangesList)
这些概念中如果使用R自带的数据结构, 可以使用如下一个数据框来表示转录本,也就是区域的向量, 每一行是一个region.
some_region <- data.frame(
seqnames= c("chr1", "chr2"),
starts = c(9932, 4393),
width = c(10923, 5428),
score= c(1, 0))
面对生物学问题时, R自带的数据结构处理起来比较麻烦.比如区域(region)的一个重要运算是判断两个region是否重叠(overlap).
我们讨厌区域之间有重叠,那么如何有重叠的区域中筛出相互不重叠的区域呢?
这些功能如果自己写代码就比较麻烦还容易出错.所以bioconductor提供了不同类型的range类来专门表示基因组中的各种区域, 比如IRange可以用来表示一维区域,Granges在IRanges的区域信息基础上提供了更多的生物学意义.
有了以上的讨论之后,bioconductor中建立IRanges和GRanges的过程就显得很自然了.
如下的代码就可以建立一个简单的IRanges对象.
IRanges(start = 101:110, end = 111:120)
而以这个IRanges对象的区域为基础,可以建立GRanges对象, 此时已经能完全确定区域的位置,并为其添加生物学信息了. 比如每个区域中碱基的GC比例.
trail_gr <- GenomicRanges::GRanges(
seqnames = rep(c("chr1", "chr2", "chr1", "chr3"), c(1, 3, 2, 4)),
ranges = IRanges(start = 101:110, end = 111:120),
strand = rep(c("-", "+", "*", "+", "-"), c(1, 2, 2, 3, 2)),
GC = seq(1, 0, length = 10)
)
与序列一一对应的数据
继续进行套娃, 染色体的序列实际上和序列的保守性信息一样, 都是一维线性有序排列的数据.
单个的染色体可以用一个很长的字符串来表示. (对应Biostrings), 同样也可以用一个数组来存储这些数值或者碱基.
然而生物相关的数据有自己的特点: 数据的取值是成片成片的. 比如说人类基因组上的编码区只占据了2%左右, 那么编码区域的突变概率直接用数组来表示就很浪费,我们不关心的非编码区占据了数组上的绝大部分下标.
这种稀疏的一维数据适合用游程编码(rle,run length encoding) 来展示:
x <- base::rev(rep(6:10,1:5))
base::rle(x)
可以看到它实际上是从头开始,逐片逐片的记录每一”片”的长度与其中数值.它用来记录编码区的突变概率的时候,编码区之间的间隔不管多长,在rle中只对应一”片”.
bioconductor 中也在base::rle的基础上提供了相应的Rle类,以及 RleList.
不同区域间的比较
在大致确定了区域 与 序列相关数据 的表示方法之后. 两者相结合就可以用来比较数据在不同区域间的差异.
然而和之前一样, “naive”的使用R原生的数据结构或者直接使用Ragnes和Rle会在”取”数据阶段耗费很多的时间. 你要去解析各个区域对应的下标, 然后到Rle选择特定的”片”……
所以bioconductor提供了Views这样的机制,用于方便的从基因组相关数据中选择感兴趣的区域并进行分析.Views相关的分析的单位是感兴趣的区域而不是数据原始的组织方式.
为了理解Views, 不妨先来看一下Cardan grille. 这是一种很古老的加密方式,它由一段文字和一个剪了很多窗口的纸片组成, 使用时将纸片覆盖到文字上,从窗口中漏出来的文字就是被加密的信息. 如下图:

同样的, 在bioconductor中间建立Views也是需要序列相关的数据以及区域. 这里序列相关的数据可以是序列本身.
比如, 如下的代码就建立了一个序列上的Views.
x <- Biostrings::DNAString("ACGTGCATCTAGCAGCGATGTCA")
mask_a <- Views(x, start = c(1, 3), end = c(3, 5))
区域也可以使用IRanges/GRanges来表示.
rl <- coverage(GRanges(seqnames = "chr1", ranges = IRanges(start = 1:10, width = 3))) # an Rle object shows coverage
grView <- GRanges("chr1", ranges = IRanges(start = 2, end = 7)) # GRanges shows the region
vi <- Views(rl, grView) # Views here
建立Views之后, 就可以用sapply来逐个窗口逐个窗口的处理数据, 也可以使用viewMaxs, viewMins, viewSums viewMeans这些自带的函数.这些函数的功能一目了然.
回到问题上来
在熟悉了bioconductor提供的这些基础设施(infrastructure)之后, 原来的问题似乎变得可解了.
大体思路就是:
- 找个办法把序列保守性数据读取进内存并转化成为Rle对象.
- 从注释文件中得到外显子在基因组上的位置信息,并转换成 GRanges对象.
- 从外显子的GRanges对象出发,得到表示外显子5’端的GRanges对象以及外显子其他区域的GRanges对象.
- 用步骤1得到的Rle对象与步骤3中得到的GRanges对象来建立Views对象.从而分析两种不同区域中数据的差异.
可以看到以上的步骤依然不完整, 只能说是大桥的桥墩已经建好,但桥墩之间的桥面还没有铺好.
接下来的部分,我们将分析这些bioconductor基础类相关的操作, 了解了这些才能把这些基础类应用起来.
全靠操作
在开始我们的讨论之前, 有必要了解一下bioconductor的结构. bioconductor是一大堆包(package)的集合,这些包的地位当然不是平等的.
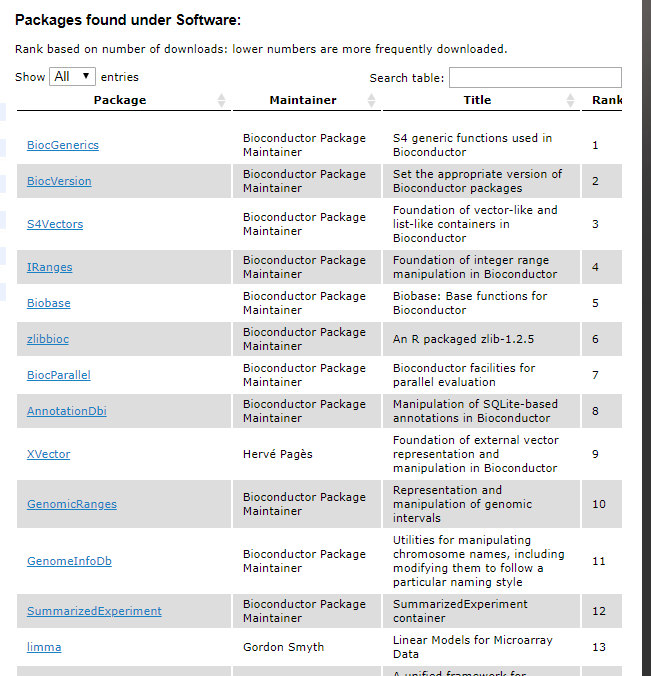
不同的包按照不同的rank排列, 这里的rank实际上就是包的重要程度, rank在前的包是后面包的基础. 可以看到上文介绍的IRanges 和GenomicRanges这两个包, 建立 GenomicRanges需要用到IRanges, 所以IRanges(rank:4)就是排在GenomicRanges(rank:10)之前的.
而排在IRanges之前, 比她还要重要/基础的包是哪些呢? 是这三个: BiocGenerics BiocVersion S4Vectors.
从包的说明可以看到, biocVersion 关注的是软件版本相关的, 这个和我们用户关系不是很大.另外两个包则相当重要:
-
BiocGenerics定义了bioconductor最通用的函数. -
S4Vectors则定义了bioconductor中向量/列表类型数据的操作, 上文中提到的Ranges/Views什么的都包括在内.
但我们不想一个猛子扎进去,直接讲解这两个包以及相关数据结构的相关函数, 因为里面内容太多, 容易扎进去浮不上来.
比如 methods(class = "IRanges")可以查看IRanges类上可以使用的函数,可以看到结果有上百个函数,我们不能按照他们函数列表的顺序来记忆这些函数.
我们更需要掌握一个”工具箱”,先想到要干什么,然后再想到什么函数可以实现它.
数据结构很多,随便一个包都可能给你提供新的数据类型,但”工具箱”中常用的顺手工具,数量并不多.
并且很多数据类型之间也是类似的.比如S4Vectors的标题中提及到’vector-like’和’list-like’,也就是说bioconductor的各种Range, RangeList都照搬了vector或者list的功能. 如果熟悉R的基本数据操作,那么稍加扩展就能理解bioconductor提供的新功能.
八字诀: 增改查删,显算传异
提到计算机科学中的数据, 常常听到这么一个”成语”:增改查删, 英文说法是 CRUD(Create, Read, Update, Delete), 指的是数据/模型应该有的最基础的功能.
如果用这个四字真言去搜索,往往能发现很多人发帖表示自己的工作很久结果工作内容就是增改查删这些没技术含量的活,因此怕被解雇.可见这些操作确实是和数据相关的最基础工作,或者说基本功.
然而越是基础的越有”迁移力”,你越会在其他地方发现它.R的数据结构相关的操作和功能自然也不例外, 并且bioconductor的数据结构基础也是R中的vector或者list. 在接下来的阐述中我们先叙述R中基本数据结构是如何实现相关的功能,然后再讨论bioconductor中特有的操作,得以观其大略.
这里我们使用”增改查删”的一个”扩展版本”,除了原来的四字真言之外,添加 显算传异.
其中的
显:数据的呈现和显示,以及所有有助于人理解数据的功能
算:与数据结构关联的常用运算.
传:数据的存贮与传输.
异:常见异常.
这八字真言原本来自于产品经理的说法,我对定义做了一些改动,但这一套走下来,可以丰富处理数据的”工具箱”, 对任何数据结构的理解都应该基本够用了.
R中数据的基础操作
使用英语作为母语的人学习计算机相关的语言是有先天优势的.因为常见计算机语言中的函数什么的都是取自英语.而那些不那么直接的函数名称往往也是缩写.
一维向量vector
R中最基础的一维数据结构就是原子向量(atomic vector), 也就是 c, seq, rep 这些函数返回的数据类型.可以用下标(index)来访问向量中特定的某个值,而且同一个向量中的值必须是同一种类型.
其他语言中的标量, 在R里面实际上就是长度为1的向量.
如下的代码可以建立简单的vector,它的数值类型是 numeric.
va <- 1:10
vb <- rep(c(1,2,3), c(2,3,4))
vector除了数据外, 可以附带一个names属性.
names(va) <- head(letters, 10)
- 增
使用 c() 或者append()可以向量中添加数据或者合并多个向量.其中 c是 concatenate的缩写
-
改 R中的修改就是重新赋值, 比如
va[1] <- 9 -
查 查询数据或者广义地说, 定位(locate)到感兴趣的数据,重要性要高于其他操作, 因为它是进行后续操作的前提. 我们要修改部分数据,首先要定位到这些数据上.
vector使用下标来定位数据, 形如a[b],其中a是向量, b就是下标.
常见的下标有三种:
- 数值 定位到对应数字下标的数据, 如果下标是负数,指的是剔除部分数据, 比如a[-1]表示剔除第一个数据.
- 布尔变量(logical)
会选中布尔向量中True对应的数据.或者使用
subset函数. - 字符 这个要求向量有names属性. R会去寻找names属性中对应的数据.
这三种不同的下标也可以互相转换, 比如使用 which来寻找布尔变量中True的下标.也可以使用R的字符处理函数来实现字符向布尔变量转换.
此外,R 提供了 head, tail, sample等等常见的选择.
- 删
R中的删除实际上就是赋值为空(NULL).
-
显 如果只是显示相关的数据, 那么直接在R的交互界面上输入相应变量的名字,然后回车就可以显示变量的内容.
如果是广义的让人更好地理解数据,那么相关的操作就比较多了. 要先确认数据比较”干净”,然后对干净的数据进行描述, 人才能更好理解面对的数据. “干净”的数据应该是没有缺失值,最好没有重复值. 另外如果可以排序的话,排好序的数据更方便之后的分析.
- 缺失值相关
使用
is.na或者is.nan之类的函数来检测数据中的缺失值. 使用na.omit来排除缺失值.
缺失值得处理是个很大的话题,可以出书的那种,这里只是希望碰到数据的时候,时刻意识到缺失值可能存在.
-
重复值 可以用
duplicated识别重复值, 或者直接unique移除重复值. -
顺序 可以用
orderrank来展示数据中的顺序情况,也可以直接用sort来得到排好序的数据.
描述性的分析(Descriptive statistical analysis)也是个很大的话题,最简单的有
meanmedianminmaxfivenumvar等等, - 缺失值相关
使用
-
算 对于一维向量来说常见的计算模式就是从头到尾挨个算一遍.这种迭代的基本形式是
for循环. R对于atomic vector上的计算有优化, 比如对向量a所有值加一,直接使用a+1比用for循环一遍快得多. 所以在R的优化上也有”向量化”这个概念,就是尽量把数据换成vector的形式以加速运算. -
传 作为普通的R语言使用者,很难碰到这方面的需求,直接使用就是.
-
异 这个部分要在使用中慢慢积累. 向量相关常见的异常,常见的是下标越界或者数据类型不一致.
IRanges/GRanges 带interval操作的向量
作为vector-like的数据结构, 可以把IRanges/GRranges理解为数据为区域region的向量(vector),